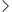原子转移自由基聚合(ATRP)是制备结构规整、分子量可控、分散度窄的 PEG-PAA(聚乙二醇 - 聚丙烯酸)嵌段共聚物的核心方法。其核心原理是通过卤原子的可逆转移实现活性 / 可控自由基聚合,避免常规自由基聚合的链终止和链转移问题。
由于聚丙烯酸(PAA)的羧基(-COOH)会与 ATRP 催化剂(铜盐)发生络合,导致聚合失控,因此实验室和工业中普遍采用 “先聚合丙烯酸酯,再水解" 的两步策略,具体合成流程如下:
PEG 的末端羟基(-OH)无法直接引发 ATRP,需先将其转化为含卤素的引发位点,以PEG-Br 的制备为例:
取一定量的单甲氧基聚乙二醇(mPEG-OH) 溶于干燥二氯甲烷(DCM),冰水浴冷却至 0~5℃,加入三乙胺(TEA)作为缚酸剂。
缓慢滴加2 - 溴异丁酰溴(摩尔比 mPEG-OH:2 - 溴异丁酰溴:TEA = 1:1.2:1.5),滴加完毕后,室温搅拌反应 12~24h。
反应结束后,过滤除去生成的三乙胺氢溴酸盐沉淀,滤液用去离子水洗涤 3~5 次,除去未反应的小分子试剂。
有机相经无水硫酸镁干燥、旋蒸浓缩后,用Ethyl ether沉淀产物,真空干燥得到mPEG-Br,通过 ¹H-NMR 表征确认引发剂制备成功(特征峰:溴异丁酰基的甲基峰~1.9 ppm)。
这一步是实现嵌段结构可控合成的关键,典型操作如下:
在史莱克管(Schlenk tube,无水无氧反应装置)中,依次加入 mPEG-Br、丙烯酸甲酯(MA)、CuBr、PMDETA,再加入干燥 THF 作为溶剂,密封反应管。
经 3 次冷冻 - 抽真空 - 解冻循环,排除管内氧气(氧气会猝灭自由基,导致聚合失败)。
将反应管置于油浴中,30~60℃恒温搅拌反应,反应时间根据目标分子量调整(通常 6~24h)。
聚合结束后,将反应液暴露于空气终止聚合,通过中性氧化铝柱除去铜催化剂,旋蒸浓缩滤液。
用正己烷 / 石油醚沉淀产物,真空干燥得到PEG-b-PMA,通过 GPC 测定分子量和分散度(PDI 通常 < 1.2),¹H-NMR 确认 MA 的聚合度。
通过碱性水解将 PEG-b-PMA 的酯基转化为羧基,得到目标产物 PEG-PAA:
将 PEG-b-PMA 溶于THF / 去离子水混合溶剂(体积比 1:1),加入过量 NaOH 水溶液(摩尔比 NaOH:MA 单元 = 3:1)。
室温搅拌反应 8~12h,期间通过薄层色谱(TLC)监测水解进程。
反应结束后,用盐酸调节溶液 pH 至 2~3,使羧基质子化。
旋蒸除去 THF,将水溶液用透析袋(截留分子量 MWCO = 1000~3000 Da)透析 24~48h,除去盐和小分子杂质。
透析液经冷冻干燥,得到白色粉末状PEG-PAA,通过酸碱滴定法测定羧基含量,确认水解。
无水无氧环境:ATRP 对氧气敏感,需严格除氧,否则会导致聚合速率降低或分子量分布变宽。
催化剂 / 配体比例:CuBr 与 PMDETA 的摩尔比通常为 1:1~1:2,配体过量可提高催化剂溶解性和聚合可控性。
单体 / 引发剂比例:通过调整 MA 与 mPEG-Br 的摩尔比,可精准调控 PMA 链段的长度,进而控制 PEG-PAA 的整体分子量。
水解条件:避免高温和过长水解时间,防止酯键过度水解导致聚合物链断裂。
聚合过程可控,可精准设计 PEG 和 PAA 链段的长度比例。
产物分散度窄,结构均一性高,适合制备高性能药物载体和生物医用材料。
可拓展制备复杂拓扑结构的 PEG-PAA(如星型、梳型共聚物)。

 热门搜索:嵌段共聚物 PEG衍生物 上转换纳米颗粒 磷脂脂质体 纳米材料 荧光染料等
热门搜索:嵌段共聚物 PEG衍生物 上转换纳米颗粒 磷脂脂质体 纳米材料 荧光染料等
 更新时间:2025-12-23
更新时间:2025-12-23 点击次数:760
点击次数:760
 当前位置:
当前位置: